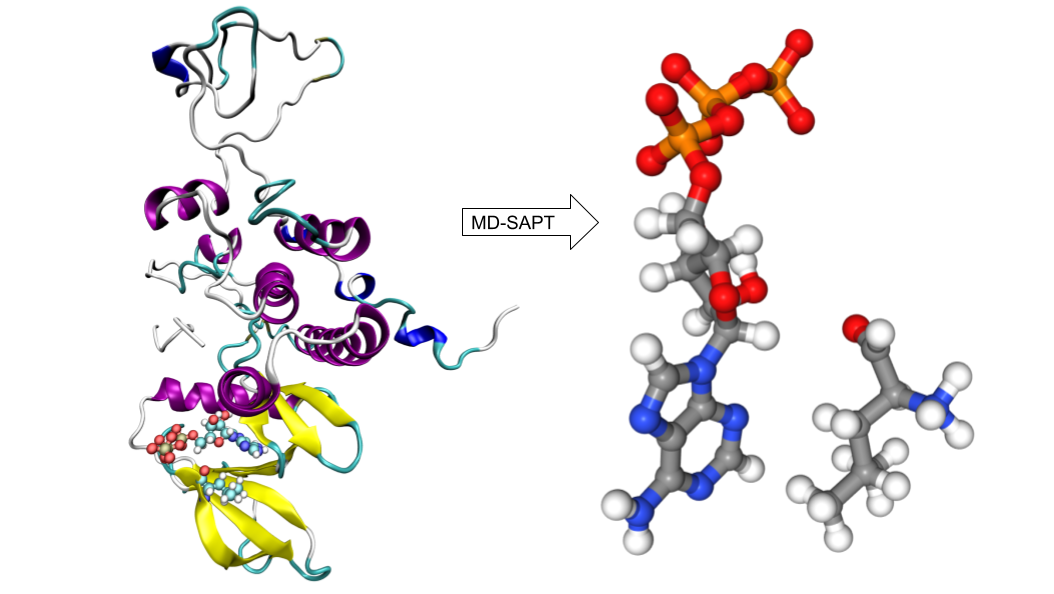
MD-SAPT
Python Based Toolkit for Running Quantum-Chemical Analysis on Molecular Dynamics Trajectories
Project Source
During the summer of 2021 I did an internship at Arizona State University in Oliver Beckstein’s computational biophysics group, during which learned about how to build software that solves scientific problems.
During that time I learned a great deal both about the fundamentals of software engineering and how to develop in the open-source Python ecosystem. Those skills were what enabled me to start MD-SAPT which went on to define my work in the Computational Chemistry Group over the subsequent years of college.
Project Summary
Molecular dynamics (MD) simulations can identify important interactions in biomolecular systems and examine how these interactions change over the course of the simulation. Biomolecular interactions modeled with MD can be also analyzed in more detail using quantum chemical methods. To accomplish this, we developed MD-SAPT, an open-source Python package, to perform quantum calculations as a post-processing analysis of MD data to quantify and decompose intermolecular interaction energy. MD-SAPT has two modes of analysis: trajectory, which analyzes selected interaction pairs over the frames of an MD simulation, and docking, to examine the binding interactions between a protein and different ligands (a small molecule that binds to a protein) or compare different docking poses for one particular ligand to determine the relative magnitude of the interactions.
Project Posts
Why I made MD-SAPT
February 4, 2024
by Alia Lescoulie
MD-SAPT is a piece of software I made for analyzing protein interactions with quantum mechanics, This post goes into the science and inspiration for this project. read more ...